Cardiac Amyloidosis for Cardiologists
- This website section is designed to provide a point of care tool for the cardiologist to assist with the recognition and diagnosis of cardiac amyloidosis with a focus on the diagnosis and management of cardiac transthyretin amyloidosis (ATTR). See ‘What is Amyloidosis?’
- ATTR is the most common type of cardiac amyloidosis
- The majority of cases of cardiac ATTR can now be diagnosed using a cardiac amyloid scintigraphy scan using bone tracers without the need for biopsy
- But biopsy is required if a monoclonal gammopathy is present as light chain amyloidosis can also show positive cardiac amyloidosis scintigraphy uptake
- Genetic testing is then required to differentiate between wild-type versus inherited ATTR
- A point of care diagnostic algorithm for the identification and typing of cardiac amyloid is discussed
- There are emerging effective ATTR disease modifying therapies
- A more detailed discussion on ATTR can be found here
Details of Cardiac Amyloidosis
Epidemiology
- The most common amyloidoses subtypes that affect the heart are ATTR followed by light chain amyloidosis (AL)
- All other types of cardiac amyloidosis are very rare
- Histologically ATTR amyloid is found in 10-25% of patients with HFpEF on autopsy studies1 and an even greater proportion of cases are detected in unselected cadaveric case series
- ATTR is divided into:
- ATTRwt (wild type) previously known as senile systemic amyloidosis
- ATTRv (inherited ATTR)
- ATTRwt is the most common form of cardiac ATTR
- ATTRwt commonly occurs in elderly males
- The male:female ratio is approximately 95:52
- There can be a long prodrome of asymptomatic heart infiltration
- Carpel tunnel syndrome (from carpel tunnel synovium ATTR deposition) precedes CCF in approximately 40%
- A significant minority of ATTR cases have an inheritable TTR gene
- 17% of 4459 ATTR patients genetically tested at the UK National Amyloidosis Centre over a 25 year period had a TTR gene mutation2. The prevalence of inherited ATTR is unknown in Australia
When to suspect cardiac ATTR amyloidosis
KEY POINT
- Unexplained left ventricular thickening on transthoracic echocardiogram (TTE) is an important “flag” for the possibility of cardiac amyloidosis
The following scenarios are when ATTR should be considered:
- Heart failure with preserved ejection fraction (HFpEF)
- Unexplained left ventricular thickening on TTE (especially when in conjunction with a low voltage ECG)
- Misleading cardiac ischaemia screening test findings (which often lead to unnecessary invasive coronary angiography in ATTR patients) with these being:
- A persistently raised troponin
- A pseudo infarct pattern on ECG (leads V1-V3)
The presence of the following features with unexplained left ventricular thickening is especially suggestive of cardiac amyloidosis:
- Right ventricular thickening
- Refractory atrial fibrillation or CHB, VT/VF, syncope
- Bilateral carpel tunnel syndrome or decompression (ATTRwt)
- Family history of heart failure especially if in association with peripheral or autonomic neuropathy (ATTRv)
In general suspecting amyloidosis when there is unexplained left ventricular thickening (whatever the clinical presentation) can greatly improve the diagnosis rate for cardiac amyloidosis.
The identification and subtyping of cardiac amyloidosis
KEY POINTS
- Cardiac amyloid bone scintigraphy can identify cardiac ATTR with 100% positive predictive value so long as there is no monoclonal gammopathy
- Cardiac amyloid bone scintigraphy can obviate the need for biopsy in the majority of ATTR cases
- But if a monoclonal gammopathy is present a biopsy is required to definitively subtype the amyloidosis because cardiac AL can also have positive cardiac amyloid bone scintigraphy uptake
- Tests to exclude a monoclonal gammopathy (all 3 must be ordered) are:
- Serum Protein Electrophoresis + immunofixation (SPEP + IFE)
- Urine Protein Electrophoresis + immunofixation (UPEP + IFE)
- Serum Free Light Chain assay
- TTR gene screening is recommended in all identified cases of ATTR
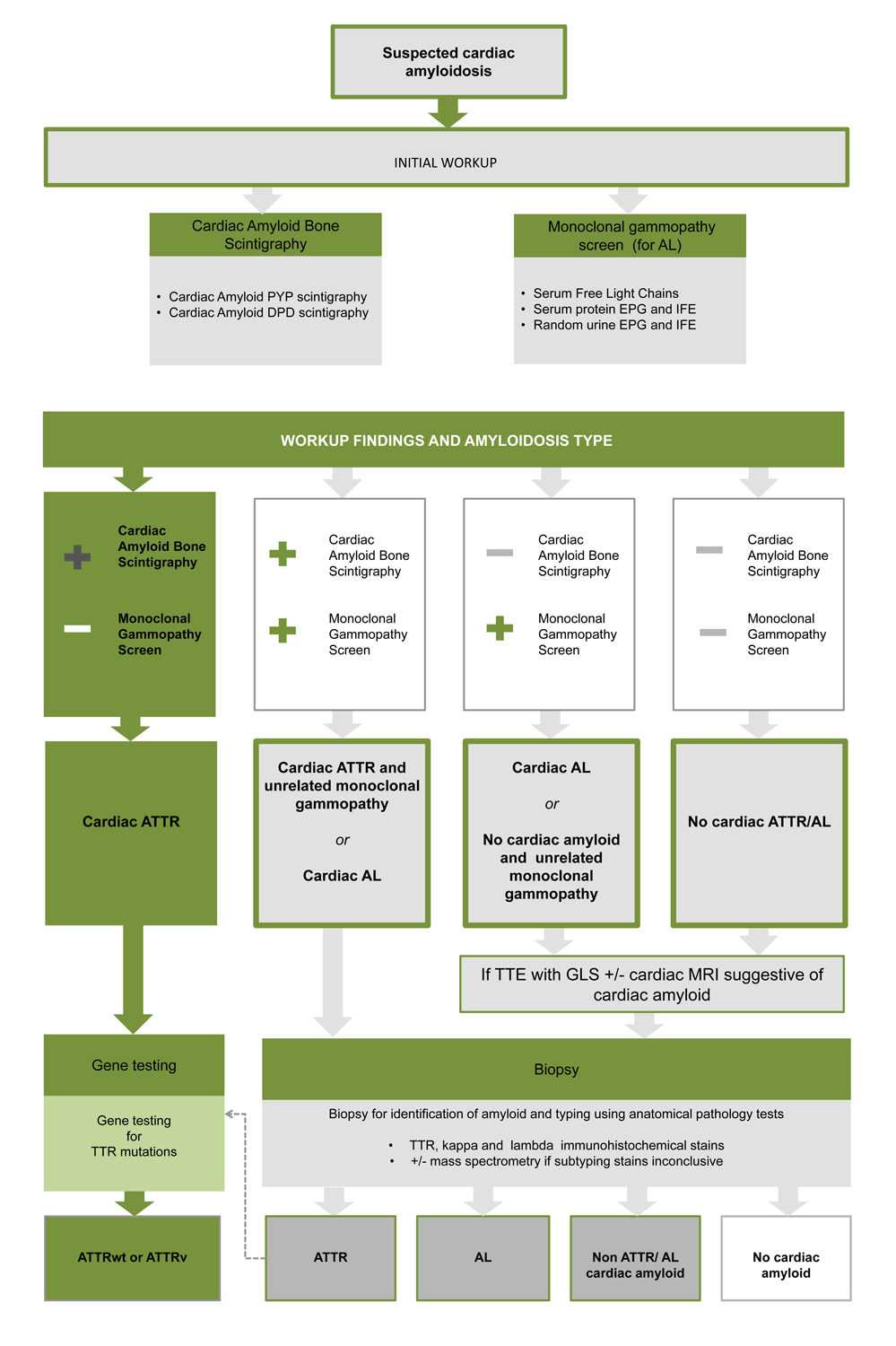
Key: PYP= pyrophosphate, DPD= 3,3 –diphosphosphono-1,2-propanodicarboxylic acid, EPG and IFE= electrophoresis and immmunoelectropheresis, ATTR= transthyretin amyloidosis, AL= light chain amyloidosis, TTE= transthoracic echocardiogram, MRI= magnetic resonance imaging, ATTRwt= wild type transthyretin amyloidosis, ATTRv= variant (aka inherited) transthyretin amyloidosis
Diagnostic algorithm explanatory notesPrognosis
- The prognosis of cardiac TTR is usually better than that for cardiac AL
- In the largest series of ATTRwt patients the median survival was 6.07 years from the onset of symptoms5
- A prognostic staging system for cardiac ATTRwt and ATTRv has been developed and validated
- This system utilises the NTproBNP and eGFR6
- Those with an NTproBNP >3000 ng/L and an eGFR <45mL/min have an estimated median OS of 24.1 months, whereas those with
- NTproBNP ≤ 3000ng/L and eGFR ≥ 45mL/min have an estimated median OS of 69.2 months.
|
Stage |
NT proBNP (ng/L)* |
eGFR (mL/min) |
Median OS (months) |
|---|---|---|---|
|
I |
≤3000 |
≥ 45mL/min |
69.2 |
|
II |
All other combinations |
All other combinations |
46.7 |
|
III |
>3000 |
<45 mL/min |
24.1 |
NTproBNP=N-terminal pro brain natriuretic peptide
eGFR= estimated glomerular filtration rate
OS=overall survival
Management of Cardiac ATTR
KEY POINTS
- Management can be divided into
- supportive care and
- disease modifying therapies
- Recently published phase III randomised controlled trials (RCT) have shown improved organ and survival outcomes for TTR stabilisers (that prevent TTR from forming amyloid) and TTR synthesis inhibitors
- The future management of ATTR is predicted to consist of a combination disease modifying therapies
Patient and Doctor Information Sheets
Diflunisal Doctor’s information (PDF)
Diflunisal Patient’s information (PDF)
Doxy and TUDCA Doctor’s information (PDF)
Doxy and TUDCA Patient’s information (PDF)
Green Tea Extract Doctor’s information (PDF)
Green Tea Extract Patient’s information (PDF)
- The management of cardiac can be divided into supportive care and disease modifying therapy
- Supportive care has been the crux of management up until now. See information on The Heart.
This centres around:- fluid management with diuretics
- careful regulation of fluid balance
- rhythm control
- ATTR disease modifying therapy can work via various mechanisms: See information on ATTR – Disease Modifying Therapy.
- stabilising TTR (so it cannot dissociate into amyloid forming monomers)
- inhibiting TTR production
- resorbing amyloid
- Effective “stabiliser” and “inhibitors of production” have recently completed phase III studies and will become an important part of therapy in the foreseeable future
- The only TTR stabiliser assessed in a RCT is Tafamidis7.
- Tafamidis is an oral TTR stabiliser
- Tafamidis showed a lower all-cause mortality than placebo (29.5% vs. 42.9%) as well as a lower rate of cardiac related hospitalisations (0.48 per year vs. 0.70 per year)
- Tafamidis is expected to become available in Australia in 2019 via clinical trials. See more on clinical trials
- PBAC are currently considering a submission for PBS listing of Tafamidis
- The most effective TTR inhibitors work via gene therapeutic techniques such as the intravenous RNA interference therapeutic agent Patirisan and the subcutaneous antisense oligonucleotide inotersen
- Both agents have demonstrated improved organ and survival outcomes in phase III trials published in 20189
- These genetic therapies are not currently available in Australia but clinical trial access is expected in 2019. See more on clinical trials
- Currently there are three TTR modifying therapies that can be obtained in Australia but none have been assessed in randomised controlled trials. See table below. These medications have:
- Less evidence for efficacy than the aforementioned therapies
- Defined side effects
- Can be prohibitive in cost as none are Medicare/PBS rebated.
- Of these EGCG is the most accessible and has the least side effects
- For a more detailed discussion on disease modifying therapies. See section on ATTR
Table 2. Currently accessible ATTR modifying therapies
|
Therapy
|
Mechanism |
Dose |
Trial type |
Number of patients
|
Efficacy |
Toxicity |
Administrative |
Cost per month* |
|---|---|---|---|---|---|---|---|---|
| Epigallaocatechin -3-gallate (EGCG)
or Green Tea Extract10 |
TTR resorber | 600mg daily | Case cohort | 25 ATTRwt | n=12/14 showed a decrease in left ventricular myocardial mass by 5.9% |
Liver function derangement (rarely liver failure)
Insomnia |
Can be purchased online | ≈$25 |
| Diflunisal11
(A NSAID) |
TTR stabiliser | 250mg tds | Phase III placebo controlled RCT | n=130 ATTRv with neuropathy | 29.7% diflunisal vs. 9.4% placebo achieved a reduced rate of neurologic progression and improved QOL |
As per NSAID side effects i.e. upper GIT side effects, renal impairment |
TGA approval required | ≈ $70 |
| Doxycycline and tauroursodeoxycholic acid (TUDCA)12 | TTR synthesis inhibitor and resorber | Doxycycline 100mg bd
TUDCA 250mg tds |
Phase II | n=40
ATTRwt and ATTRv cases |
Stable neuropathy observed in n=6/13 evaluable patients. n=18/24 evaluable patients had stable NTproBNP |
Doxycycline side effects are mainly sun sensitivity skin reactions, GIT side effects |
TGA approval required for TUDCA | Doxycycline can be PBS funded for certain indications
TUDCA≈$115 |
Key:
ATTR=transthyretin amyloidosis
TTR= transthyretin
ATTRwt= wild type transthyretin amyloidosis
NSAID= non-steroidal anti-inflammatory drug
RCT= randomized controlled trial
ATTRv= variant (inherited) transthyretin amyloidosis
QOL= quality of life
TGA= Therapeutic Goods Administration
*Please note the cost per month only pertains to the cost of the medication and this varies depending on the pharmaceutical importer. This cost does not include pharmacist handling fee or GST. These costs were last updated April 2019.