AL Amyloidosis
AL amyloidosis is the most commonly diagnosed type of amyloidosis in the Western world.
- It is not inherited or contagious.
- It is acquired, meaning it just happens.
- It is an extremely complicated systemic disease which varies in presentation from person to person.
- Usually more than one organ is involved.
- No one knows why some people develop AL amyloidosis while others do not.
- Without treatment the prognosis is poor, especially when heart function is impaired by the amyloid deposition.
Prognosis
Over the last 20 years with improved awareness, earlier diagnosis and better techniques to diagnose, type and treat AL amyloidosis, many people are achieving a disease remission and living normal lives. However, AL amyloidosis remains a complicated disease that without treatment often has a very poor outcome, particularly if the heart is involved or the disease is diagnosed late. Early diagnosis is still the key to more effective treatment.
What causes AL amyloidosis?
Normal plasma cells, antibodies and free light chains
Before talking about what causes AL amyloidosis, first a few words about the normal situation in the human body.
Plasma cells are a special kind of white blood cell which are part of the body’s immune system. Plasma cells live in the bone marrow and their job is to make antibodies which are also known as immunoglobulins. Antibodies are a special type of protein and are part of the body’s defense system against infection. The human body is very adaptable and can make thousands of different antibodies in order to deal with many different infections. Once antibodies have done their job they are broken down and recycled in the body.
Each antibody molecule (or immunoglobulin) is made of two “heavy” chains and two “light” chains joined together (see diagram at right). The light chains can be of two types, Kappa or Lambda. When the immune system makes antibodies, light chains are made in excess of the amount needed to produce an antibody. These excess light chains circulate in the blood as free light chains.. Normally everyone has small amounts of kappa and lambda free light chains in their blood. These light chains circulate in the blood, cause no harm, and are excreted in the urine.
Plasma cells diseases and amyloid forming free light chains
In AL amyloidosis, plasma cells in the bone marrow begin to proliferate or grow abnormally, a situation known as a plasma cell disorder. Usually this buildup of plasma cells is benign but in some cases the growth of plasma cells can be malignant, that is, a cancer of the bone marrow called multiple myeloma. These abnormal plasma cells make large amounts of a single type of free light chain. The free light chain proteins fold in an abnormal way, become amyloid fibrils and progressively deposit in any of the tissues and organs of the body, except the brain. This deposition slowly interferes with organ and tissue function. Without treatment to slow or stop this amyloid being produced and deposited, organ failure occurs.

| Plasma cell proliferation | Does the free light chain form amyloid? | Disease |
|---|---|---|
| Cancerous (malignant) | No | Myeloma |
| Cancerous | Yes | Myeloma + AL amyloidosis |
| Non-malignant | No | MGUS (monoclonal gammopathy of undetermined significance) |
| Non-malignant | Yes | AL Amyloidosis |
Organ Involvement
- AL amyloidosis is a complex systemic disease, meaning that any tissues of the body except the brain can be affected by the deposition of the amyloid protein.
- There is no common pattern and great variation between patients in the way the amyloid protein is laid down.
- In most patients more than one organ will be affected.
The most common organs affected are:
- Kidneys
- Heart
- Nerves, with both autonomic and peripheral neuropathy.
- Liver
- Gastrointestinal tract.
Symptoms
Symptoms may be vague and nonspecific and often mimic other diseases.
- Loss of appetite
- Tiredness
- Loss of weight
- Weakness
More specific symptoms will depend on which organs are affected by the amyloid deposition.
- Swollen ankles (oedema) – Kidney or heart involvement
- Breathlessness, especially on effort and going up a slope – Heart involvement
- Tingling in the fingers and toes – Nerve involvement
- Frothy urine – Kidney involvement
- Bruising, especially around the eyes (raccoon eyes) – Amyloid in the blood vessels
- High cholesterol – Kidney involvement
- Diarrhea and constipation – Gut or autonomic nerve involvement
- Swollen tongue (macroglossia) – Tongue involvement
Diagnosing AL Amyloidosis
Early diagnosis is essential but often very difficult. Unfortunately, many patients visit a number of doctors before the diagnosis is made and then present with severe organ damage, which can compromise treatment.
No single blood test exists to diagnose amyloidosis.
Diagnosis begins when a doctor suspects the patient’s symptoms may be due to amyloidosis and starts to run tests or makes a referral to a specialist with knowledge in treating amyloidosis.
AL amyloidosis can only be definitively diagnosed through a tissue or organ biopsy.
- A tissue biopsy involves removing a small piece of tissue for microscopic examination from the symptomatic organ thought to be involved by amyloid or from a distant site such as the fat pad on the tummy, the gastrointestinal tract, the lip or the bone marrow.
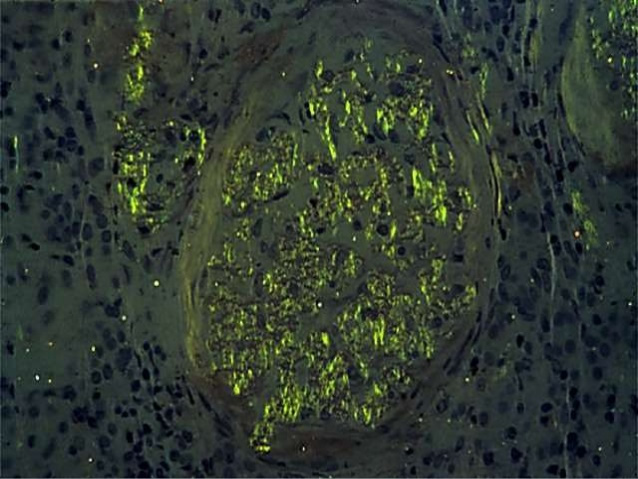
- The biopsy is stained with the dye Congo red in the laboratory.
- If amyloid is present, the biopsy will appear red under normal light and green (apple-green birefringence) under special polarized light.
- Further testing may be undertaken on the biopsy to identify the type of amyloidosis. This is important, as the treatment for each type of amyloidosis is different.
- However in the case of a patient being diagnosed with amyloidosis and macroglossia (swelling of the tongue) this testing will not be needed as this is only seen in AL amyloidosis.
See section on amyloidosis for information on different types.
A series of further tests will be run to:
- Identify which organs are affected by the amyloid deposition and to what degree.
- Check the general health of the patient including other health problems that may affect treatment.
- Help with typing the disease.
- To enable a treatment plan to be made.
Tests may include:
- A number of blood tests to amongst other things: kidney (creatinine), liver (liver function tests) and heart function (serum troponin and BNP or NT-proBNP)
- Echocardiogram
- ECG
- CT scan, MRI
- Genetic testing may be performed to exclude one of the hereditary types of amyloidosis.
- A bone marrow biopsy to establish the presence of abnormal plasma cells
- Urine analysis to measure the amount of protein and any light chains in the urine (Bence Jones proteinuria)
- Free Light Chain assay and serum protein electrophoresis
It is usually only when all these results have been collated and consideration has been given to:
- Patients age
- Where they live
- Whether a treatment trial is available and appropriate
that a treatment plan will be recommended in consultation with the patient.
Treatment
Clinical Practise Guidelines

Professor Morie Gertz: What is AL Amyloidosis?
Future AL amyloidosis treatment

AL amyloidosis treatment including stem cell transplantation – Mayo Clinic