AA Amyloidosis
Amyloid A (AA) amyloidosis is the third most common type of systemic amyloidosis in Australia. However, the majority still remain predominantly of ATTR or AL type with AA cases only constituting 3-5% of systemic amyloidosis cases referred to the AAN clinics1.
Serum amyloid A (SAA) is the constituent protein in AA amyloid deposits and is an acute phase inflammatory protein. AA amyloidosis is strongly associated with chronic inflammation. The most common underlying inflammatory disorders in the “First World” are the chronic inflammatory arthritides followed by the “idiopathic” group then infectious diseases2. In up to 20%-27% of cases, no inflammatory cause is found. Obesity is an emerging associated condition in this “idiopathic” group3.
Details of the AA Amyloid type
Epidemiology
- In Australia, AA amyloidosis is the third most common form of systemic amyloidosis but it still only constitutes a minority of systemic cases
- AA comprises 3-5% of systemic cases referred to the AAN clinics, with ATTR and AL constituting approximately 95% of all systemic amyloidosis cases1
- This reflects advances in therapy for autoimmune disorders and chronic infections
- AA comprises 3-5% of systemic cases referred to the AAN clinics, with ATTR and AL constituting approximately 95% of all systemic amyloidosis cases1
- Inflammatory arthritides are the most common group of disorders that cause AA amyloidosis, followed by the “idiopathic” group, then chronic infections2
- Obesity is an emerging associated condition in the idiopathic group
- The “obesity epidemic” may change the epidemiology of AA in the future
Pathophysiology
- The amyloid forming protein in AA amyloid is Serum Amyloid A
- SAA is an inflammatory protein that belongs to the pentraxin family of proteins
- The C-reactive protein belongs to the same family of proteins
- SAA is an apolipoprotein of high-density lipoproteins that serves as a dynamic acute phase reactant
- It is a significant part of the “acute phase response”
- The level of SAA increases more dramatically than that of any other protein and may reach 2,000 mg/L3
- It is synthesized as a precursor by hepatocytes in response to transcriptional stimuli from various proinflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumour necrosis factor (TNF) alpha
- Two different isoforms of SAA have been isolated and SAA1 is responsible for most of the serum elevation during the acute phase response
- It is a significant part of the “acute phase response”
- The factors involved in the molecular kinetics of AA amyloid formation are not fully understood but a sustained and high level of SAA is required and there are also predisposing genetic factors
- The median duration of the inflammatory disorder prior to diagnosis is 17 years but it may be as short as one month2
- Certain genetic mutations can predispose to AA amyloidosis:
- MEFV mutations
- especially MEFV M694V (the most common mutation in familial Mediterranean fever)
- SAA genetic factors regulate susceptibility to the deposition of AA amyloid
- The SAA genotype is an important determinant of amyloidogenesis in certain inflammatory conditions but ethnic differences have been observed
- SAA1 is the precursor amyloidogenic protein in most individuals with AA amyloid
- SAA1 alleles are risk factors for the development of AA amyloidosis particularly when there is homozygosity for certain alleles5
- SAA1 has three alleles, designated SAA1.1, SAA1.3, and SAA1.5
- For Caucasians with rheumatoid arthritis, the presence of the SAA1.1 allele is associated with a higher risk of developing AA amyloidosis5
- In Japanese patients, homozygosity for the SAA1.3 allele is associated with a greater increase in SAA levels, a shorter latency period before disease onset, more severe systemic damage, and shorter survival than other allelic variants6
- The SAA genotype is an important determinant of amyloidogenesis in certain inflammatory conditions but ethnic differences have been observed
- MEFV mutations
Inflammatory disorders associated with AA amyloidosis
- Any chronic inflammatory disorder can cause AA amyloid but in particular inflammatory disorders where the acute phase response is active and sustained
- In the Third World chronic infections such as TB predominate as the cause of AA amyloidosis
- In the First World the epidemiology of the inflammatory disorders causing AA amyloidosis is different
- The categories from the most common to the least common inflammatory disorders in the First World are2:
- Autoimmune disorders
- Especially rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, juvenile inflammatory arthritis and Chrohn’s Disease
- SLE is not a common cause of AA amyloidosis as the acute phase response of SAA is blunted despite the persistent inflammation
- Especially rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, juvenile inflammatory arthritis and Chrohn’s Disease
- Idiopathic
- Approximately 20-27% % of AA amyloidosis cases are idiopathic7
- AA amyloidosis of unknown aetiology rose from 10 to 27 percent of new cases in a recent epidemiologic study7
- Obesity is increasingly found in the “idiopathic” group in association with findings suggesting low-grade but chronic inflammation probably due to cytokine production by adipocytes3
- Autoinflammatory disorders
- Periodic fever syndromes such as familial Mediterranean fever, Cryopyrin-associated periodic syndromes, Muckle-Wells syndrome and TNF-receptor-associated periodic fever syndrome (TRAPS)
- Chronic infection/sepsis
- Bronchiectasis, paraplegia (with chronic infection of pressure sores and urinary retention)2 and in the context of intravenous drug use
- AA amyloidosis was the most frequent cause of kidney disease in 50% of intravenous drug users evaluated at two different reference centres in Germany and was associated with chronic HIV infection and a previous history of repeated skin and soft tissue infections8
- Bronchiectasis, paraplegia (with chronic infection of pressure sores and urinary retention)2 and in the context of intravenous drug use
- Malignancies
- renal cell cancer, lymphoma, mesothelioma
- Other
- Castleman’s syndrome, IgG4 disorder, vasculitides
- Any disorder that is associated with the acute phase response in a sustained manner can potentially cause AA amyloidosis and there are many case reports that cover a broad range of associated inflammatory disorders
Pattern of organ involvement
- The most common sites of clinical organ disease are2:
- Kidneys ≈ 97%
- GIT ≈ 10%
- Liver ≈ 10%
- Almost all patients (>95%) have renal involvement and present with proteinuria:
- Proteinuria is often in the nephrotic range
- The median protein excretion at diagnosis (for those not requiring dialysis) is approximately 4g/day2
- Renal impairment
- At diagnosis approximately 10% are in end-stage renal failure
- But for those who present with a creatinine clearance of >20mL/min the median creatinine level is 99umol/L2
- Proteinuria is often in the nephrotic range
Diagram 1. AA clinical organ distribution2. Showing the symptomatic or clinically detectable sites of organ involvement.
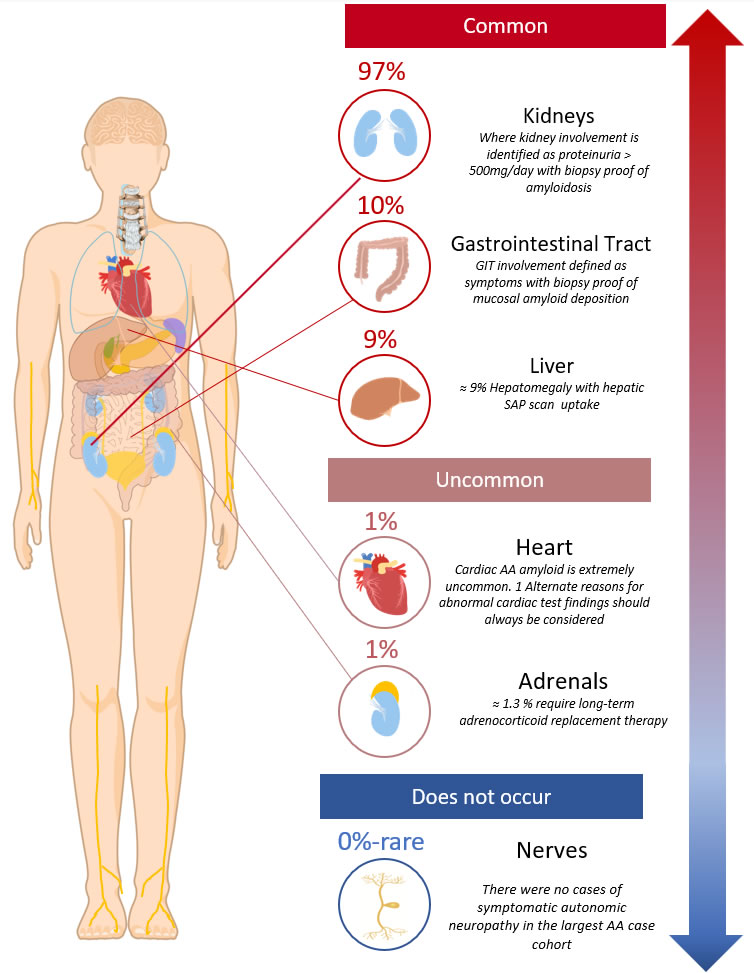
- It is important to note that the heart is rarely involved in AA amyloidosis (only in ≈ 1%2) and that there has only been the rare case report of AA amyloid related neuropathy (affecting the autonomic nervous system 9,10)
- Asymptomatic AA amyloid deposits (which do not cause clinical signs or symptoms) are detectable by SAP scintigraphy in organs at a greater frequency than is clinically evident2 i.e:
- spleen 99%
- adrenals 41%
- liver 23%
Diagnosis and basic workup
- The diagnosis of AA amyloidosis requires the combination of;
- Amyloid typing
- tissue based amyloid typing tests
- screening for other types of renal amyloidosis
- AL is the most common type of renal amyloid
- Rare types of renal amyloidosis are inherited renal amyloidosis (AFib) and LECT2 See “the kidney”
- organ staging
- searching for the underlying inflammatory process
- Amyloid typing
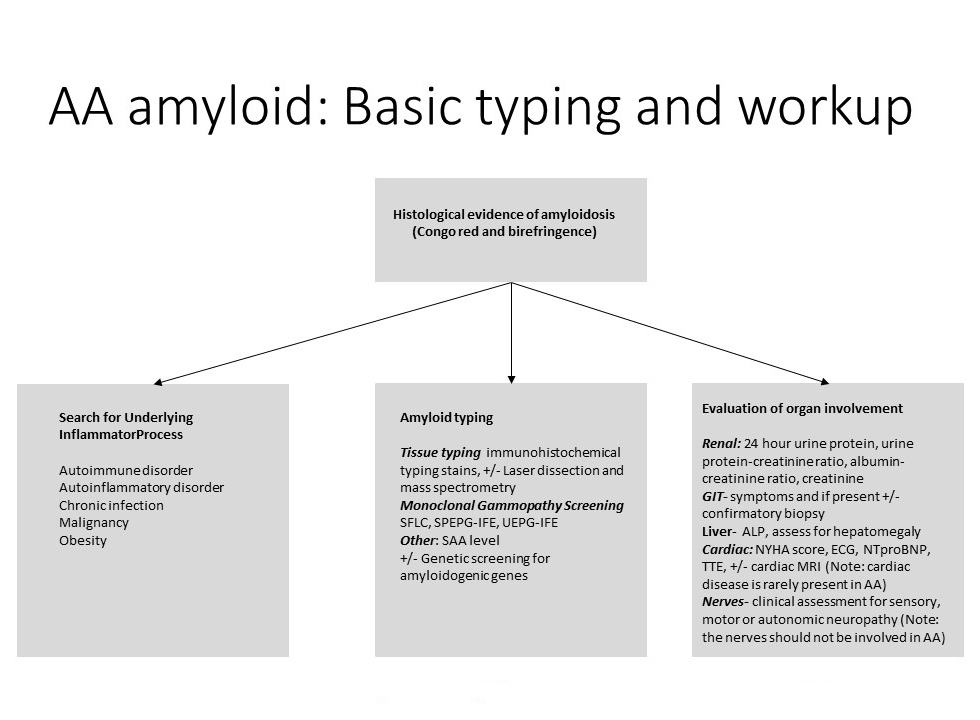
Amyloid typing
- A tissue biopsy is always required to identify the presence of amyloid and to perform amyloid typing tests upon See “What is amyloidosis”
- Histopathologic typing of the amyloid using a panel of immunohistochemical stains including the AA stain is mandatory
- Unfortunately, subtyping immunohistochemical stains are subject to false positive (and less commonly false negative) AA staining results and cannot be used in isolation to type the amyloid deposits TO ADD: Link to basic overview of diagnosis and tissue typing
- Histopathologic typing of the amyloid using a panel of immunohistochemical stains including the AA stain is mandatory
- Screening for other types of amyloid and in particular for AL amyloid is required TO ADD: link Basics: Amyloid typing
- This requires serum free light chain(SLFC), serum protein electrophoresis and immunofixation (SPEPG-IFE), 24-hour urine for urine electrophoresis and immunofixation (UEPG-IFE)
- Approximately 98% of AL cases will have a raised monoclonal amyloidogenic FLC (TO ADD: see AL amyloid; clonal gammopathy findings)
- All three screening tests will detect the presence of an AL plasma cell clone in approximately 99% of AL cases12
- The presence of an unrelated monoclonal gammopathy may confound the amyloid typing process
- If typing tests or organ distribution are inconsistent then the following typing tests are suggested:
- Laser capture microdissection and mass spectrometry of the amyloid from the paraffin embedded tissue block
- Gene screening for amyloidogenic mutations
Organ staging
- Organ screening is recommended as part of the diagnostic workup TO ADD: link Basic overview: organ staging
- If there is heart or nerve disease, alternate types of systemic amyloidosis (especially AL) need to be considered and excluded
- In summary organ screening tests consist of:
- Renal: 24-hour urine protein quantitation/urine protein-creatinine ratio, albumin-creatinine ratio, creatinine
- GIT: upper and lower GIT symptoms and if present +/- confirmatory GIT biopsy
- Liver: hepatomegaly, ALP
- Spleen: blood film for hyposplenic features
- Cardiac: NT-proBNP, troponin, +/- TTE
- Nerves: clinical examination +/- nerve studies
Identifying the inflammatory disorder
- It is usually clearly evident if the underlying inflammatory disorder is an autoimmune disease or chronic infection
- In cryptogenic cases the following can be considered;
- CT +/- PET scan for occult malignancy
- Testing for inherited periodic fever syndromes
Prognosis
- The median survival in the largest cohort analysis of AA amyloidosis patients was 133 months after diagnosis2
- Survival and renal outcomes were correlated with SAA concentration
- The lower the SAA the better the overall survival and renal outcome
- Amyloid deposits regressed in 60% of cases where the median SAA <10 mg/L2
- A SAA level of < 10mg/L therefore correlates with no AA amyloid production (or a clinically insignificant amount of AA amyloid formation)
- Amyloid deposits can regress if the production of AA is stopped or reduced enough for a patient’s endogenous macrophage clearance mechanisms to clear amyloid
- Not all patients have the ability of endogenously clear amyloid
- This process is slow
- Amyloid deposits can regress if the production of AA is stopped or reduced enough for a patient’s endogenous macrophage clearance mechanisms to clear amyloid
- A SAA level of < 10mg/L therefore correlates with no AA amyloid production (or a clinically insignificant amount of AA amyloid formation)
- Aiming for the lowest possible level of SAA is also associated with improved survival
- This survival effect is even seen by comparing different target levels within the “normal” SAA reference range. In Lachmann et al’s case series2
- With the reference range for SAA being <6 mg/L, a median SAA of
- <4mg/L was associated with a relative risk of death of 1.0
- ≥4 to <9mg/L was associated with a relative risk of death of 3.9
- Hence, ideally it is best to maintain a median SAA in the low as possible and even into the lowest part of the “normal” reference range
- With the reference range for SAA being <6 mg/L, a median SAA of
- This survival effect is even seen by comparing different target levels within the “normal” SAA reference range. In Lachmann et al’s case series2
Treatment
- The treatment goal is to prevent endstage renal failure, improve overall survival and increase the chances of renal recovery (by allowing endogenous amyloid clearance)
- Supportive care is also very important and revolves around supporting the failing kidney
- Treatment is directed at suppressing the underlying inflammatory disorder i.e:
- Disease modifying therapies in Rheumatoid Arthritis associated AA
- Chemotherapy in lymphoma related cases etc
- Weight loss is suggested for those where obesity is the thought to be an inflammatory factor
- In idiopathic cases empiric “anti-inflammasome” therapy includes:
- Colchicine
- Steroids
- Anakinra
- Tocilizumab
- The most commonly used empiric therapy is colchicine:
- The therapeutic dose is 500 micrograms three times a day but patients may achieve therapeutic efficacy at lower doses
- Diarrhoea is often a dose-limiting side effect and anti-diarrhoeal agents such as Immodium may be required
- It is advised to treat infections promptly for any patient with AA amyloidosis (of any underlying cause) as infection may provoke an additional surge in SAA levels followed by rapid AA deposition (a so-called “AA storm”)
Monitoring
- It is suggested to aim for a SAA level of <10 mg/L
- And if possible, even for a SAA level in the lower part of the reference range i.e. <4mg/L
- Regular SAA levels are recommended (See Lab Contacts) at a monthly frequency especially when therapy is being instituted, then less frequently when control of SAA levels is achieved.
- The CRP is usually not as sensitive as a biomarker of AA amyloid production but it can correlate well with the SAA in some cases and the lab assay is more accessible than the SAA
- Consider performing paired CRP and SAA levels when initiating therapy to determine the utility of using the CRP as a longterm surrogate surveillance biomarker of AA production
- Regular urine protein assessments (with protein creatinine and albumin creatinine ratios +/- 24-hour urine protein quantitation) can determine if there is amyloid clearance after a low SAA level is achieved
- Renal recovery may occur quickly over several months after a SAA of <10mg/L is reached but it may take up to 18 months to occur